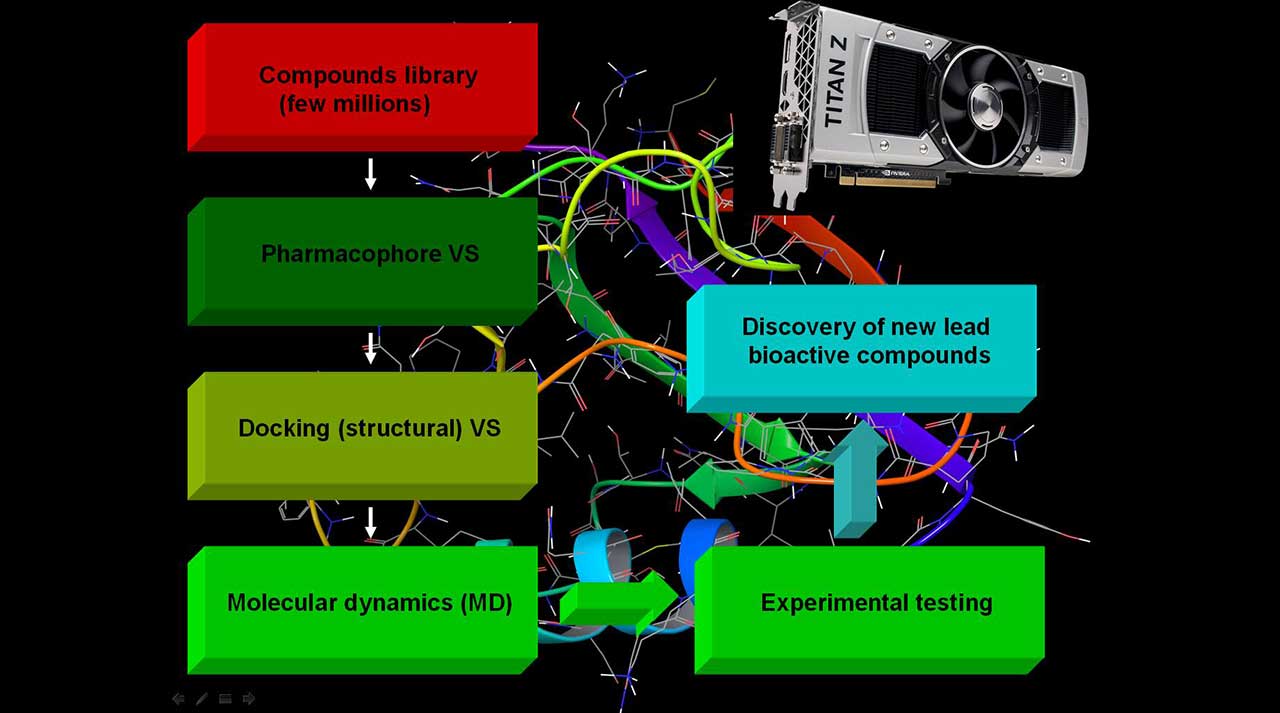
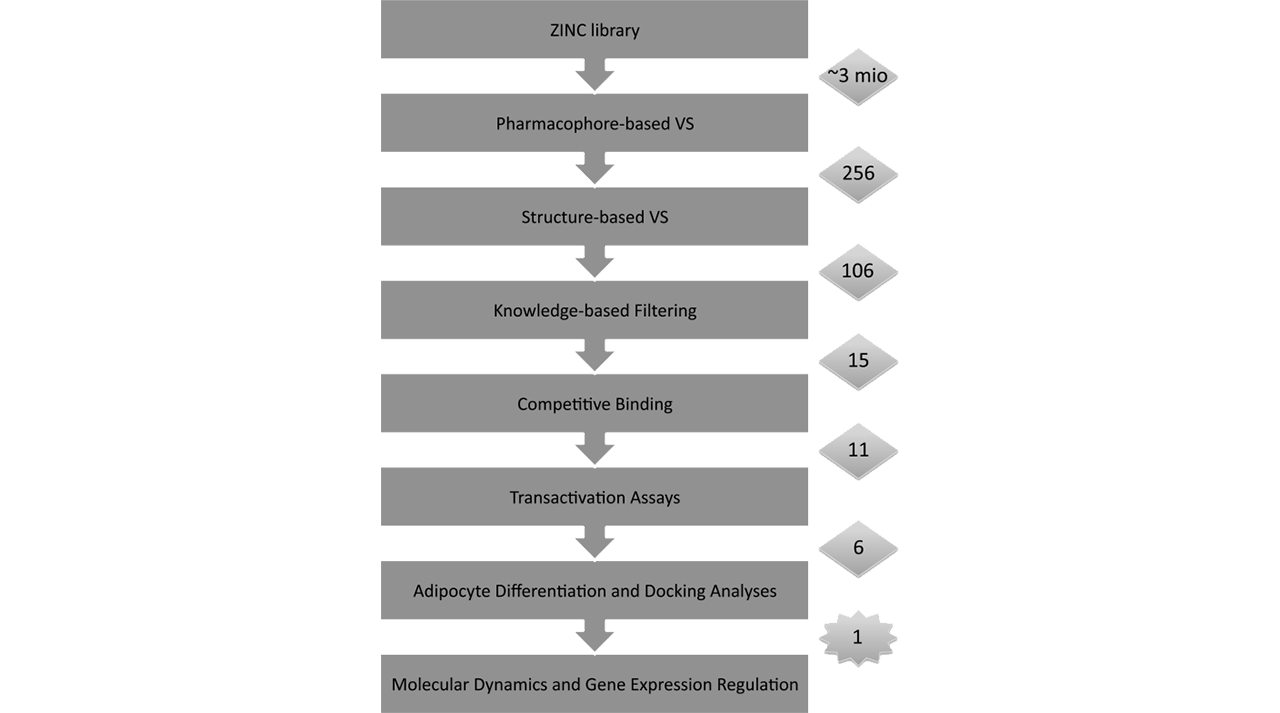
IN SILICO DRUG DESIGN
HIT IDENTIFICATION
$
9 999
*
per project
- * + part of IP
- in vitro test not included
- Using Traditional in silico
- Using power of AI
IN SILICO DRUG DESIGN
LEAD OPTIMIZATION
$
9 999
*
per project
- * + part of IP
- in vitro test not included
- ($95 per 1 permutation)
- Using Traditional in silico
- Using power of AI
IN SILICO DRUG DESIGN
CHANGES IN THE LIGAND BINDING AFFINITY AND SELECTIVITY INTRODUCED BY POINT MUTATIONS
$
5 999
*
per project
- * + part of IP
- ($165 per 1 mutation)
- Using Traditional in silico
- Using power of AI
IN SILICO DRUG DESIGN
RECEPTOR STRUCTURE GENERATION AND REFINEMENT
$
5 999
per project
- Using Traditional in silico
- Using power of AI